
Irzam Sarfraz, Yichen Wang, Amulya Shastry, Wei Kheng Teh, Artem Sokolov, Brian R. Herb, Heather H. Creasy, Isaac Virshup, Ruben Dries, Kylee Degatano, Anup Mahurkar, Daniel J. Schnell, Pedro Madrigal, Jason Hilton, Nils Gehlenborg, Timothy Tickle & Joshua D. Campbell Genome Biology volume 25, Article number: 205 (2024) Cite this article 236 Accesses 5 Altmetric Metricsdetails Abstract Many datasets are being produced by consortia that seek to characterize healthy and disease tissues at single-cell resolution. While biospecimen and experimental information ……
Category Archives: josh_pub

05
Nov
Nov
Aaron Chevalier # 1 2, Tao Guo # 1 3, Natasha Q Gurevich # 1 2, Jingwen Xu 1 3, Masanao Yajima 3, Joshua D Campbell 1 2 Affiliations Expand Abstract The majority of mutational signatures have been characterized in tumors from American and European countries, and the degree to which mutational signatures are similar to or different from those in Chinese populations has not been fully explored. We leveraged a large-scale clinical sequencing cohort ……
04
Sep
Sep
Yuan Yin 1, Masanao Yajima 2, Joshua D Campbell 1 Affiliations Expand Abstract Assays such as CITE-seq can measure the abundance of cell surface proteins on individual cells using antibody derived tags (ADTs). However, many ADTs have high levels of background noise that can obfuscate down-stream analyses. In an exploratory analysis of PBMC datasets, we find that some droplets that were ……

11
Oct
Oct
Chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD) are clinically and molecularly heterogeneous diseases. We utilized clustering and integrative network analyses to elucidate roles for microRNAs (miRNAs) and miRNA isoforms (isomiRs) in COPD and ILD pathogenesis. Short RNA sequencing was performed on 351 lung tissue samples of COPD (n=145), ILD (n=144) and controls ……

11
Oct
Oct
Analysis of single-cell RNA-seq (scRNA-seq) data can reveal novel insights into heterogeneity of complex biological systems. Many tools and workflows have been developed to perform different types of analysis. However, these tools are spread across different packages or programming environments, rely on different underlying data structures, and can only be utilized by people with knowledge ……
11
Oct
Oct
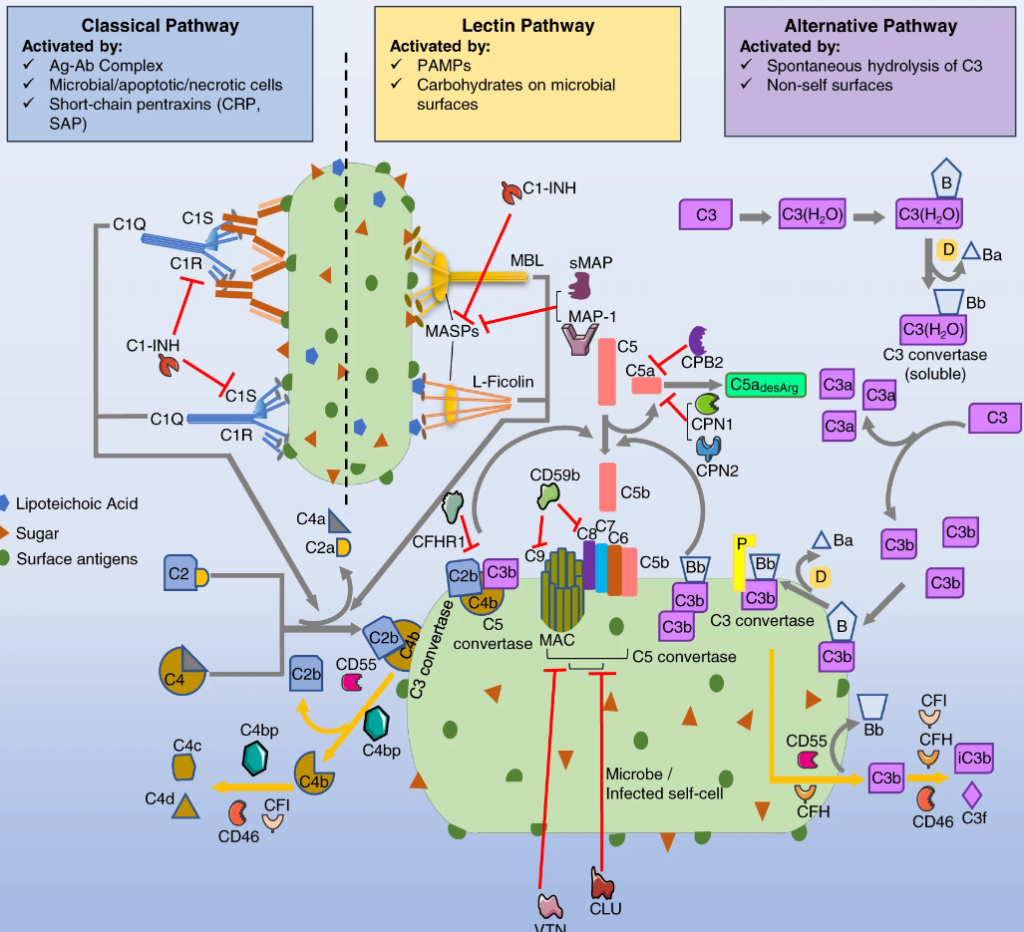
To understand functional duality of the complement system in host defense and lung injury, a more comprehensive view of its localized production in the lung, and the impact of age on complement production are essential. Here, we explored the expression of complement genes through computational analysis of preexisting single cell RNA sequencing data from lung ……
11
Oct
Oct
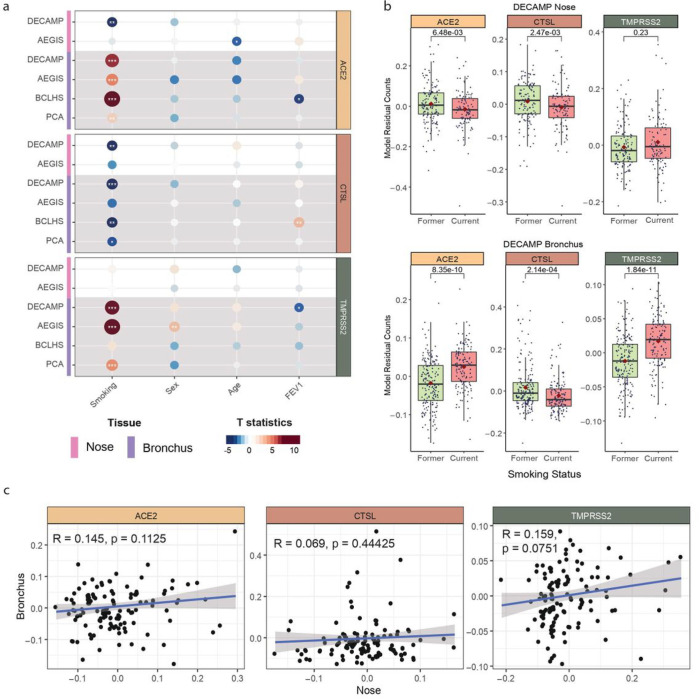
Xu, K., Shi, X., Husted, C., Hong, R., Wang, Y., Ning, B., Sullivan, T., Rieger-Christ, K., Duan, F., Marques, H., Gower, A., Xiao, X., Liu, H., Liu, G., Duclos, G., Platt, M., Spira, A., Mazzilli, S., Billatos, E., Lenburg, M., … Beane, J. (accepted by scientific report) Abstract Background: SARS-CoV-2 infection and disease severity are ……
11
Oct
Oct
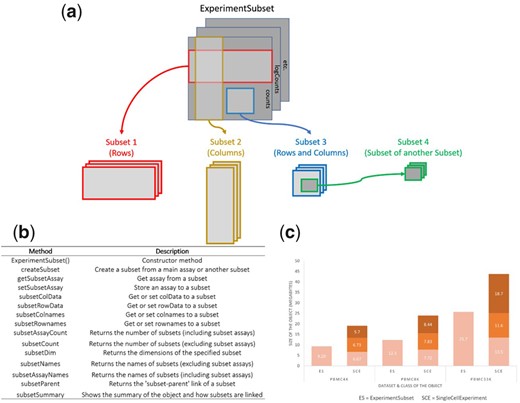
Motivation R Experiment objects such as the SummarizedExperiment or SingleCellExperiment are data containers for storing one or more matrix-like assays along with associated row and column data. These objects have been used to facilitate the storage and analysis of high-throughput genomic data generated from technologies such as single-cell RNA sequencing. One common computational task in many genomics analysis workflows ……
11
Oct
Oct
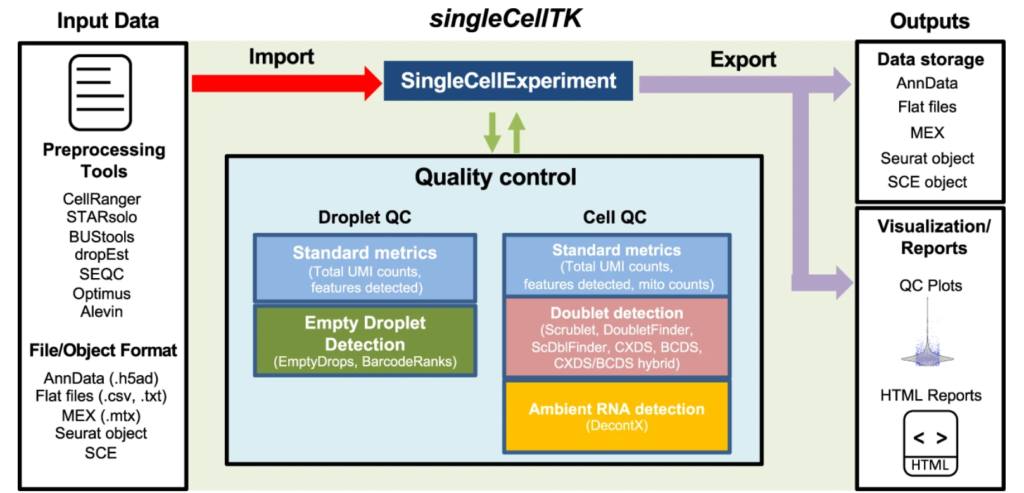
Single-cell RNA sequencing (scRNA-seq) can be used to gain insights into cellular heterogeneity within complex tissues. However, various technical artifacts can be present in scRNA-seq data and should be assessed before performing downstream analyses. While several tools have been developed to perform individual quality control (QC) tasks, they are scattered in different packages across several ……
11
Oct
Oct
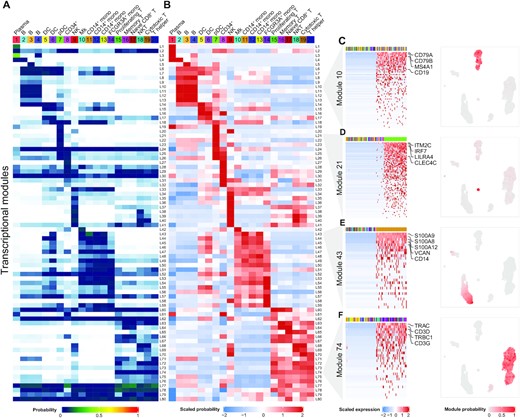
Single-cell RNA-seq (scRNA-seq) has emerged as a powerful technique to quantify gene expression in individual cells and to elucidate the molecular and cellular building blocks of complex tissues. We developed a novel Bayesian hierarchical model called Cellular Latent Dirichlet Allocation (Celda) to perform co-clustering of genes into transcriptional modules and cells into subpopulations. Celda can ……