
Irzam Sarfraz, Yichen Wang, Amulya Shastry, Wei Kheng Teh, Artem Sokolov, Brian R. Herb, Heather H. Creasy, Isaac Virshup, Ruben Dries, Kylee Degatano, Anup Mahurkar, Daniel J. Schnell, Pedro Madrigal, Jason Hilton, Nils Gehlenborg, Timothy Tickle & Joshua D. Campbell
Genome Biology volume 25, Article number: 205 (2024) Cite this article
- 236 Accesses
- 5 Altmetric
- Metricsdetails
Abstract
Many datasets are being produced by consortia that seek to characterize healthy and disease tissues at single-cell resolution. While biospecimen and experimental information is often captured, detailed metadata standards related to data matrices and analysis workflows are currently lacking. To address this, we develop the matrix and analysis metadata standards (MAMS) to serve as a resource for data centers, repositories, and tool developers. We define metadata fields for matrices and parameters commonly utilized in analytical workflows and developed the rmams package to extract MAMS from single-cell objects. Overall, MAMS promotes the harmonization, integration, and reproducibility of single-cell data across platforms.
Background
The past two decades have seen a rapid expansion of high throughput genomic and imaging technologies that have revolutionized the ability of researchers to capture the molecular and histological characteristics of biological samples. For example, assays such as single-cell RNA-seq can capture the states of individual cells within heterogeneous and complex tissues. Several major consortia have been funded to utilize single-cell assays to create cellular atlases of healthy and disease tissues [1,2,3,4,5,6,7]. These groups are generating large datasets that contain multi-modal single-cell data collected from longitudinally and spatially related biological specimens from different organs across different conditions. The majority of datasets are designed to answer a specific set of questions within a particular biological or clinical context. Other data repositories such as NIH Gene Expression Omnibus (GEO) and ArrayExpress/BioStudies databases are focused on systematic storage, cataloging, and retrieval of primary data [8, 9]. As more data becomes available from consortia and individual investigators, the ability to combine and integrate datasets from different settings is becoming increasingly desirable. Three major roadblocks to combining and integrating datasets are that (1) the metadata related to clinical, biospecimen, and experimental parameters is not captured or harmonized across groups [10]; (2) the data is stored in a wide variety of file formats or programming language-specific libraries, classes, or data structures; and (3) the metadata about the matrix and the corresponding analysis that produced or utilized the matrix is not well standardized. While significant effort has been dedicated to defining metadata standards for experimental parameters [10] and new file formats are under active development [11], the third area remains largely unaddressed.
While a wide range of experimental protocols and platforms are available to generate molecular and histological data, an important commonality across these technologies is that they often produce a matrix of features that are measured in a set of observations. These feature and observation matrices (FOMs) are foundational for storing raw data from molecular assays (e.g., raw counts) and derived data from downstream analytical tools (e.g., normalized matrix, reduced dimensional matrix). A variety of file formats are used to store FOMs on file systems in different representations. For example, tab-separated value (tsv/txt) files can be used to store dense matrices while market exchange (.mtx) files can be used to efficiently store sparse matrices. Although platform-independent, these formats do not readily capture relationships between matrices and annotations for features and observations. Several packages also exist that can capture relationships between matrices and annotations include AnnData and MUON in Python [12, 13], the Seurat object in R [14, 15], and the SingleCellExperiment and related packages in R/Bioconductor [16,17,18]. In contrast to the simple flat file formats, these objects can capture complex relationships between FOMs as well as annotation data produced during the analysis such as quality control (QC metrics) and cluster labels. However, each package may store different sets of data or label the same type of data differently. Even if different datasets are stored the same format or type of object, harmonization of datasets still may require substantial manual curation before they can be combined and integrated.
Lastly, a major goal of high-quality analysis workflows is to promote reproducibility by storing information related to provenance such as software version, function calls, and selected parameters that produced the matrix or annotation. However, there is a high degree of variability in which different analytical tools and software packages capture this information. Even if the data was produced by a workflow captured in a Docker container or versioned in GitHub, this information will often be lost when converting the data between formats or transferring between tools. Thus, there is a need to develop metadata standards for FOMs related to provenance to ensure this information can be readily captured and maintained throughout the dataset-specific analysis and during integration with other datasets.
In order to facilitate sharing of data across groups and technologies as well as to promote reproducibility related to data provenance, a detailed metadata schema describing the characteristics of FOMs can be used to serve as a standard for the community. Therefore, we developed the matrix and analysis metadata standards (MAMS) to capture the relevant information about the data matrices and annotations that are produced during common and complex analysis workflows for single-cell data. MAMS defines fields that describe what type of data is contained within a matrix, relationships between matrices, and provenance related to the tool or algorithm that created the matrix. In contrast to the existing standards, MAMS does not largely focus on information related to sample preparation including biospecimen and clinical metadata or metadata related to experimental protocols. We have also built a new R package, rmams, that automatically extracts available MAMS annotations from existing single-cell objects and stores them in a platform-agnostic file format. These standards will serve as a roadmap for tool developers and data curators to ensure that their systems have the capability to store and retrieve relevant information needed for integration. All of the metadata fields are independent of the platform, programming language, and specific tool and thus can be used to support efforts to harmonize data across consortia.
Results
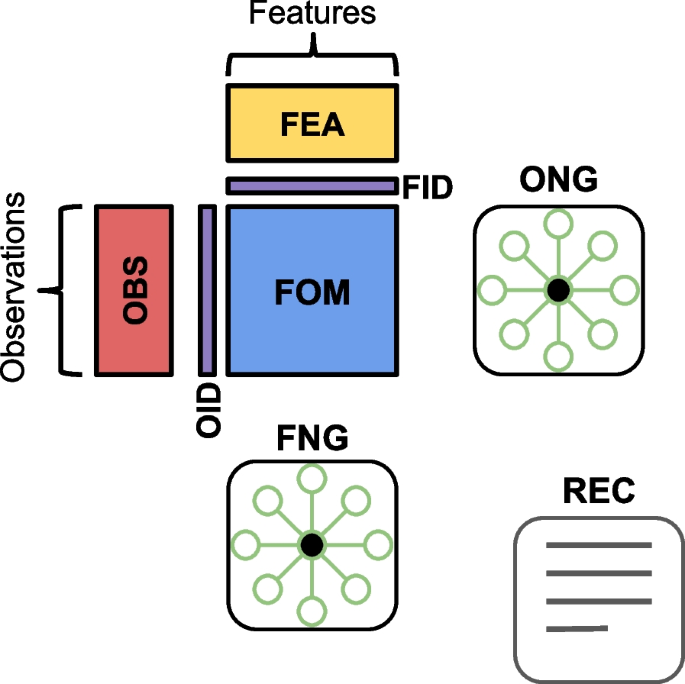
Overview of matrix classes
Several popular libraries and software packages offer convenient interfaces for storing and retrieving data matrices and their associated annotations. The majority of these tools employ similar schemas that organize different classes of data matrices in an intuitive framework with a common interface (Fig. 1). In general, we refer to a feature and observation matrix (FOM) as a class of data matrix that contains measurements of features across biological entities. Examples of features include genes, genomic regions, peaks, transcripts, proteins, antibodies derived tags, signal intensities, cell type counts, or morphology categories. Examples of observations include cells, cell pools, beads, spots, subcellular regions, and regions of interest (ROIs). Measurements for single-cell data may include transcript counts, protein abundances, signal intensities and velocity estimates. FOMs that contain raw, normalized, transformed, or standardized biological data are commonly referred to as “assays” or “layers.” In the MAMS nomenclature, FOMs can also contain reduced dimensional objects such as principal components from PCA or 2-D embedding from tSNE or UMAPs which are derived from the original biological data matrices. We note that although the term “matrix” is used in the acronym, FOMs may also be data frames which can contain mixed data types (e.g., continuous and categorical morphological features), vectors (e.g., a matrix with 1 dimension), and multidimensional arrays (e.g., a matrix with more than 2 dimensions also known as a tensor).
Open Journal to continue reading.