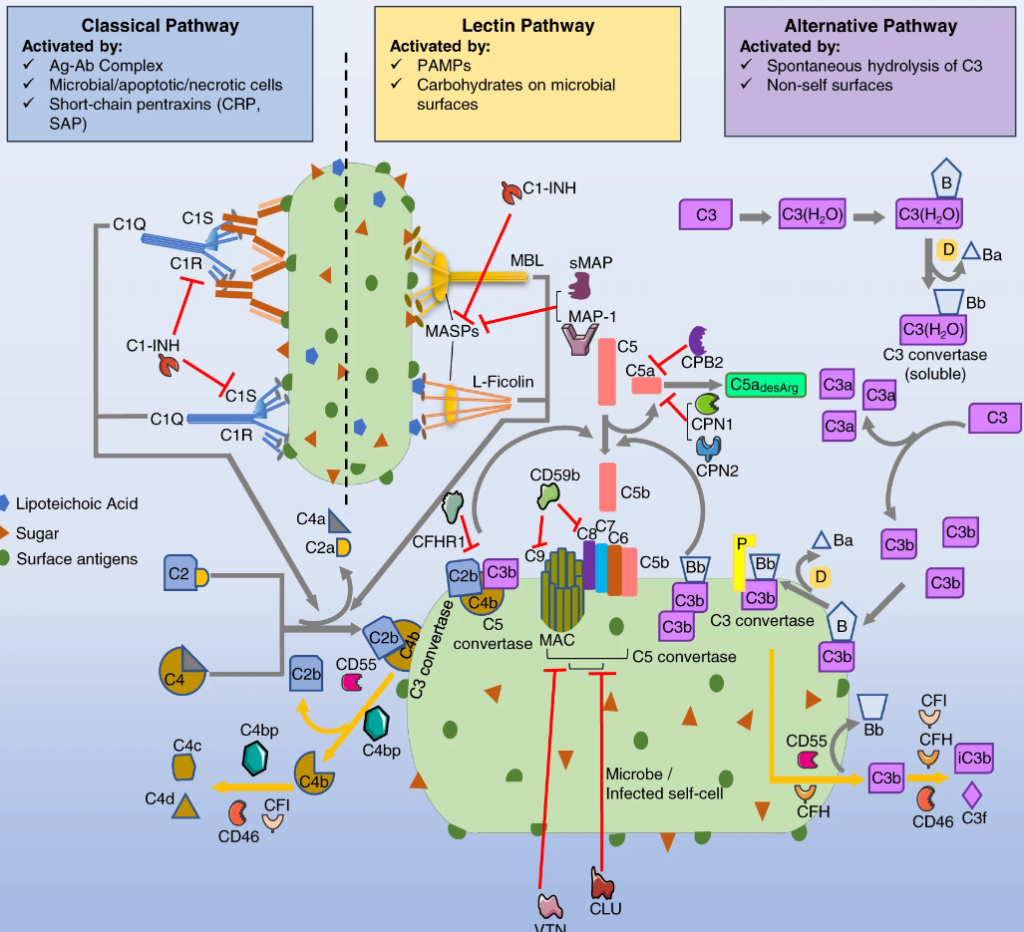
To understand functional duality of the complement system in host defense and lung injury, a more comprehensive view of its localized production in the lung, and the impact of age on complement production are essential. Here, we explored the expression of complement genes through computational analysis of preexisting single cell RNA sequencing data from lung ……
Category Archives: pub_2022
11
Oct
Oct
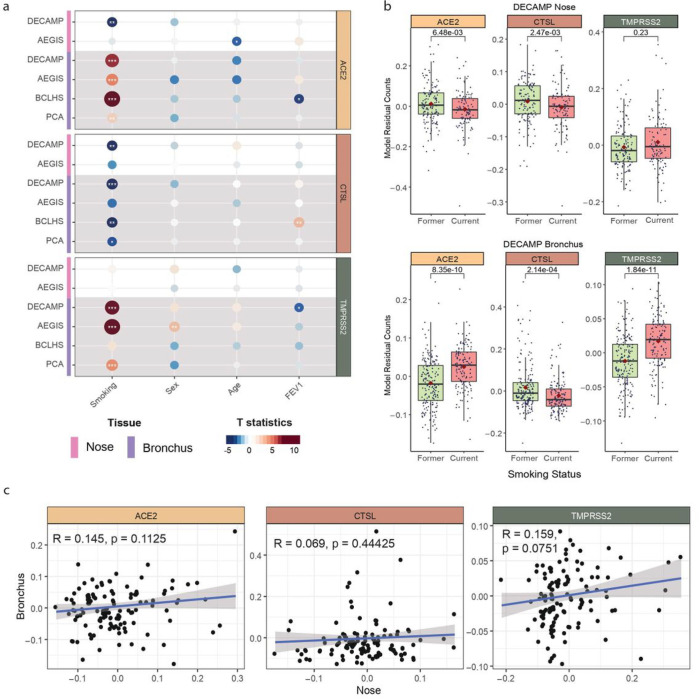
Xu, K., Shi, X., Husted, C., Hong, R., Wang, Y., Ning, B., Sullivan, T., Rieger-Christ, K., Duan, F., Marques, H., Gower, A., Xiao, X., Liu, H., Liu, G., Duclos, G., Platt, M., Spira, A., Mazzilli, S., Billatos, E., Lenburg, M., … Beane, J. (accepted by scientific report) Abstract Background: SARS-CoV-2 infection and disease severity are ……
11
Oct
Oct
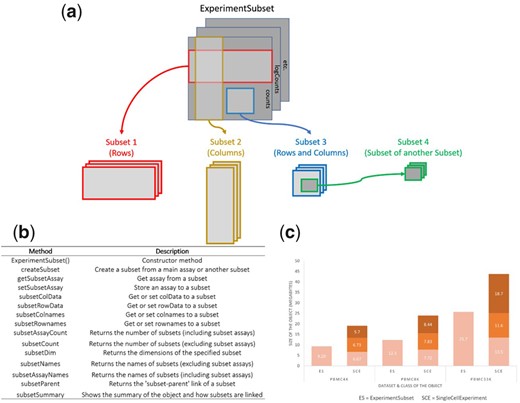
Motivation R Experiment objects such as the SummarizedExperiment or SingleCellExperiment are data containers for storing one or more matrix-like assays along with associated row and column data. These objects have been used to facilitate the storage and analysis of high-throughput genomic data generated from technologies such as single-cell RNA sequencing. One common computational task in many genomics analysis workflows ……
11
Oct
Oct
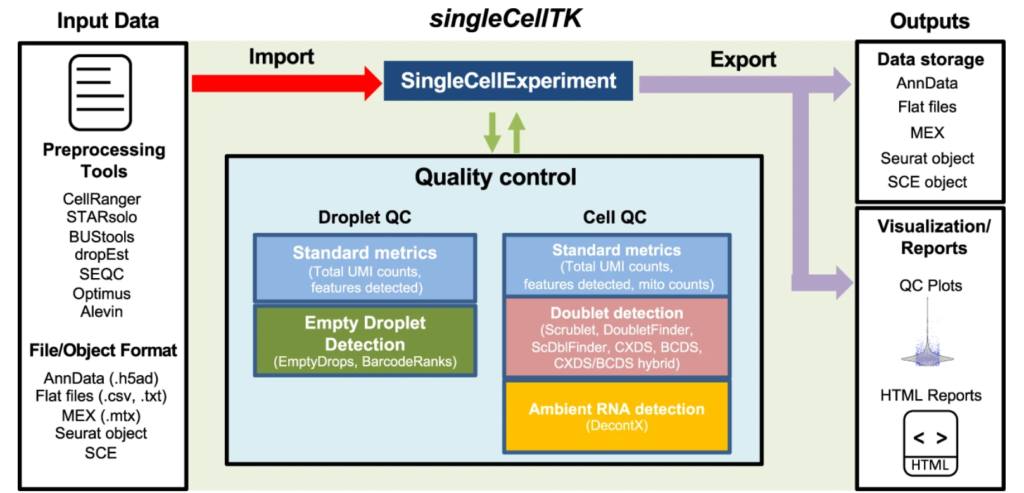
Single-cell RNA sequencing (scRNA-seq) can be used to gain insights into cellular heterogeneity within complex tissues. However, various technical artifacts can be present in scRNA-seq data and should be assessed before performing downstream analyses. While several tools have been developed to perform individual quality control (QC) tasks, they are scattered in different packages across several ……
11
Oct
Oct
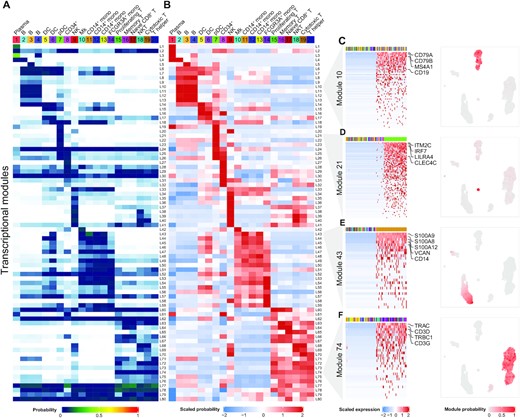
Single-cell RNA-seq (scRNA-seq) has emerged as a powerful technique to quantify gene expression in individual cells and to elucidate the molecular and cellular building blocks of complex tissues. We developed a novel Bayesian hierarchical model called Cellular Latent Dirichlet Allocation (Celda) to perform co-clustering of genes into transcriptional modules and cells into subpopulations. Celda can ……
11
Oct
Oct
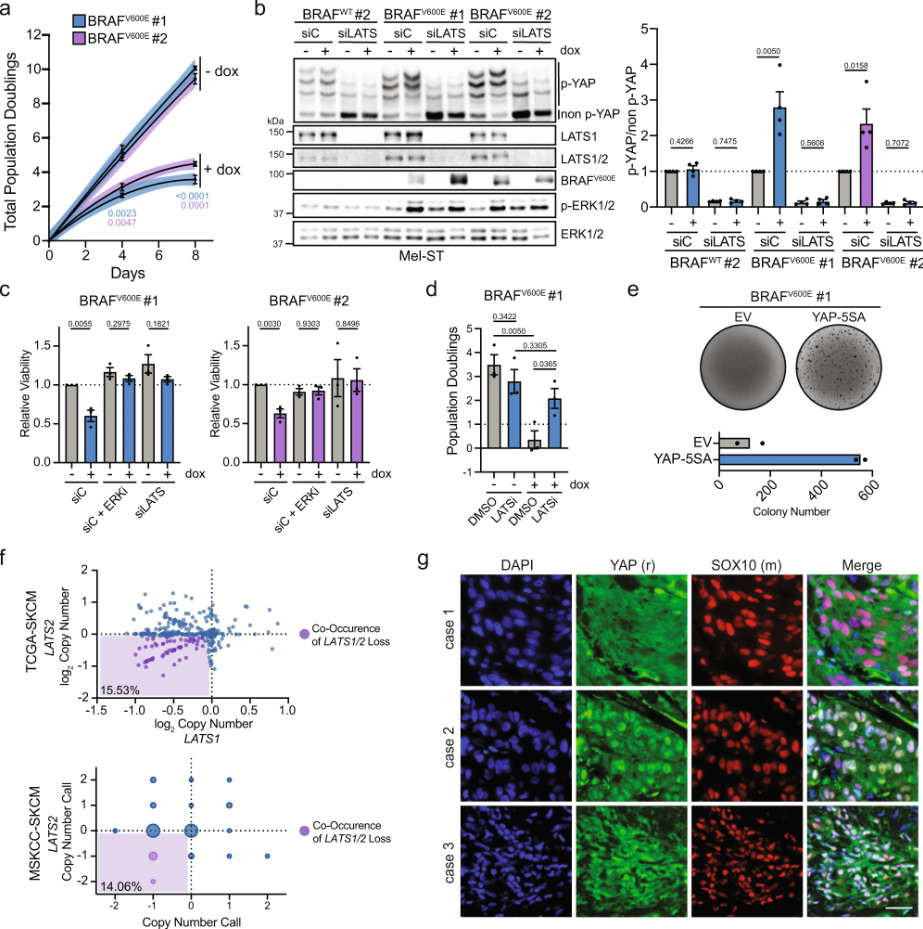
Melanoma is commonly driven by activating mutations in the MAP kinase BRAF; however, oncogenic BRAF alone is insufficient to promote melanomagenesis. Instead, its expression induces a transient proliferative burst that ultimately ceases with the development of benign nevi comprised of growth-arrested melanocytes. The tumor suppressive mechanisms that restrain nevus melanocyte proliferation remain poorly understood. Here ……
11
Oct
Oct
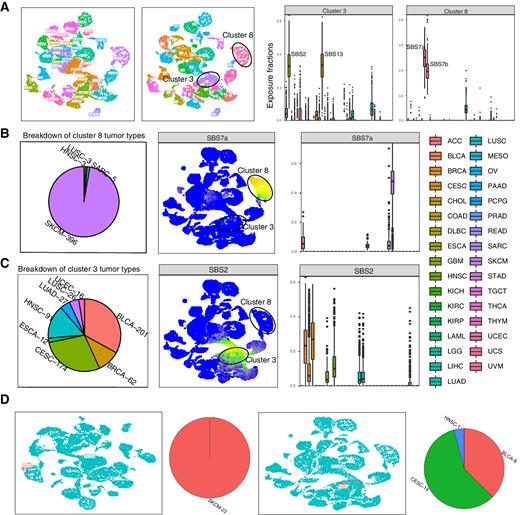
Mutational signatures are patterns of somatic alterations in the genome caused by carcinogenic exposures or aberrant cellular processes. To provide a comprehensive workflow for preprocessing, analysis, and visualization of mutational signatures, we created the Mutational Signature Comprehensive Analysis Toolkit (musicatk) package. musicatk enables users to select different schemas for counting mutation types and to easily ……