Introduction
singleCellTK offers a convenient way to normalize
data for downstream analysis using a number of methods available through
the toolkit. The integrated normalization methods and other
transformation options can be used through both R console and
interactive shiny application. Normalization methods available with the
toolkit include "LogNormalize", "CLR",
"RC" & "SCTransform" from
Seurat [[1]][2][[3]][4] package and "logNormCounts" or
"CPM" from Scater [5] package. Additional transformation options
are available for users including ‘log’ & ‘log1p’, trimming of data
assays and Z-Score scaling.
To view detailed instructions on how to use these methods, please select ‘Interactive Analysis’ for using normalization in shiny application or ‘Console Analysis’ for using these methods on R console from the tabs below:
Workflow Guide
Normalization tab can be opened up by clicking on the Normalization & Batch Correction from the top menu and further selecting the Normalization sub-tab in the subsequent window as shown below.

Normalization tab is divided up into 4 sections as
shown in the screenshot below:
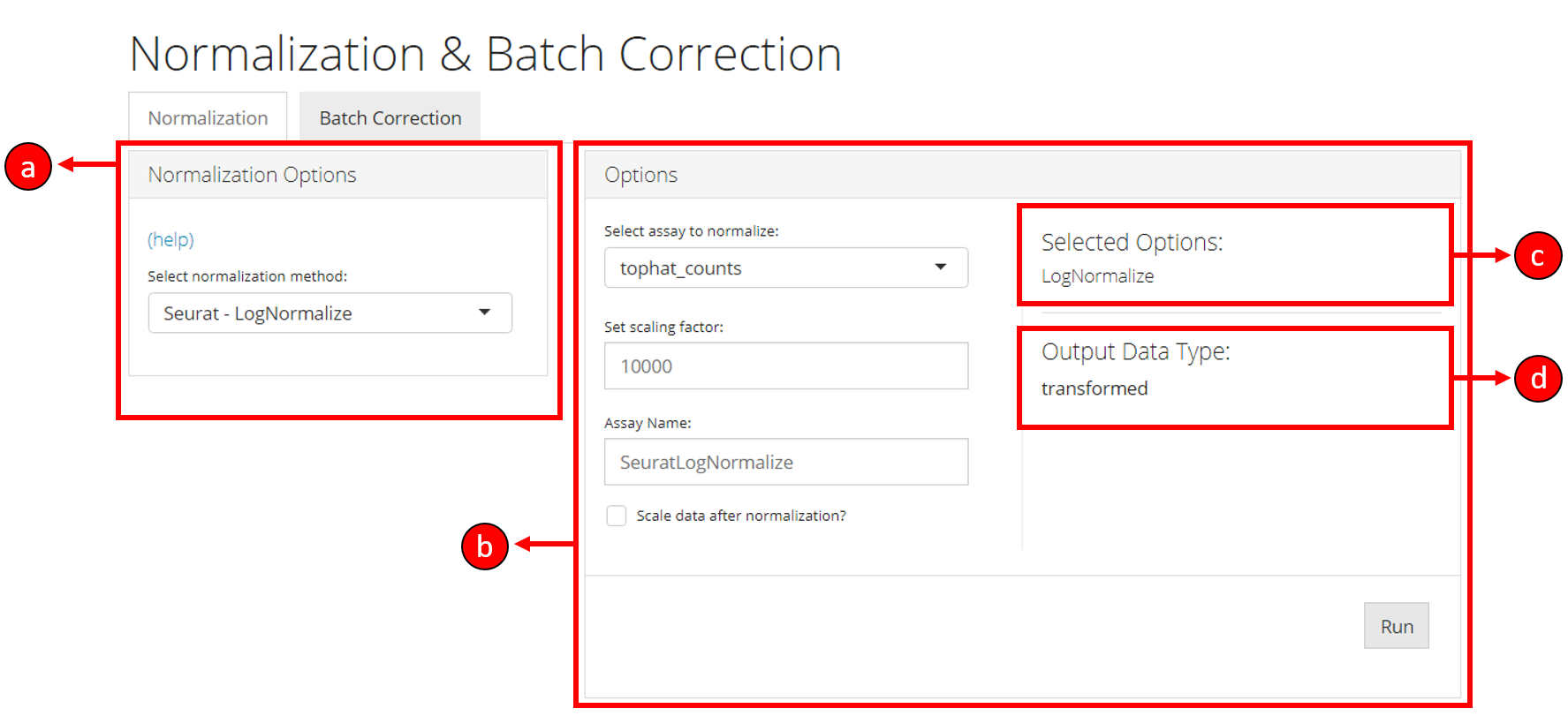
The section a lets you choose a normalization method,
whereas the section b allows for various options to be
modified and selected for a particular normalization method selected
from section a. The section c displays
the overall methods/options selected for this instance of normalization
process. The section d displays the output data type
once the normalization is run. This output data type represents the
nature of the data that will be output based on the selected options.
For example, output data type maybe transformed if
transformation options are selected including normalization methods, or
maybe scaled if the scale option is
selected. The section c and d update
automatically based on normalization method and options selected by the
user.
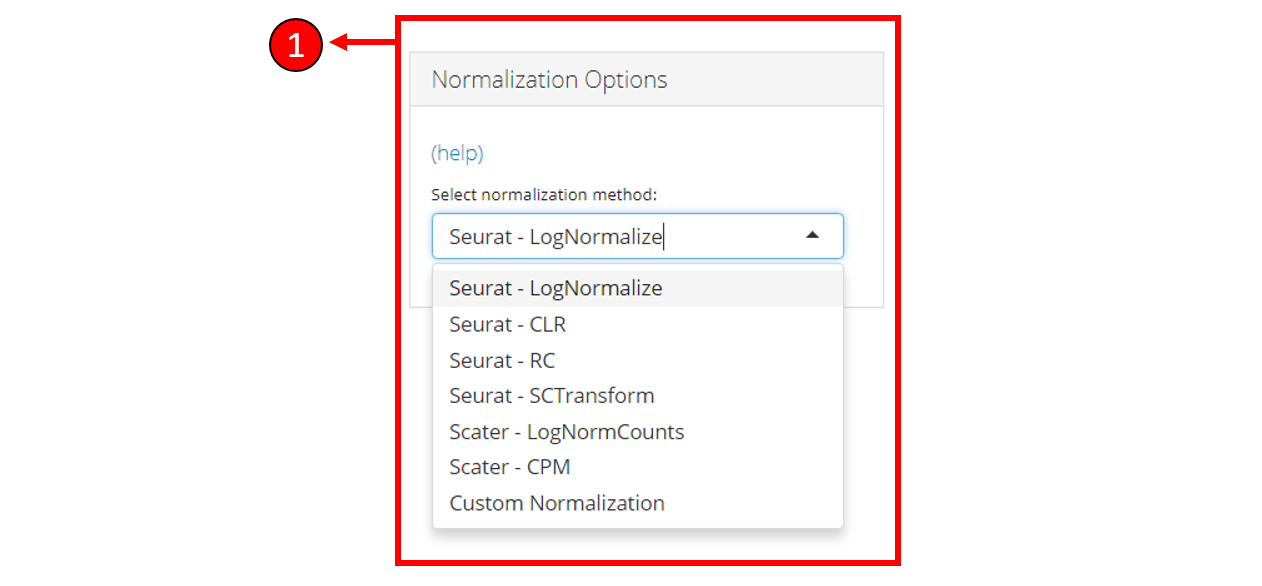
1. Select a normalization method or select
Custom Normalization to manually select the options for
normalization including transformation, trimming & scaling.
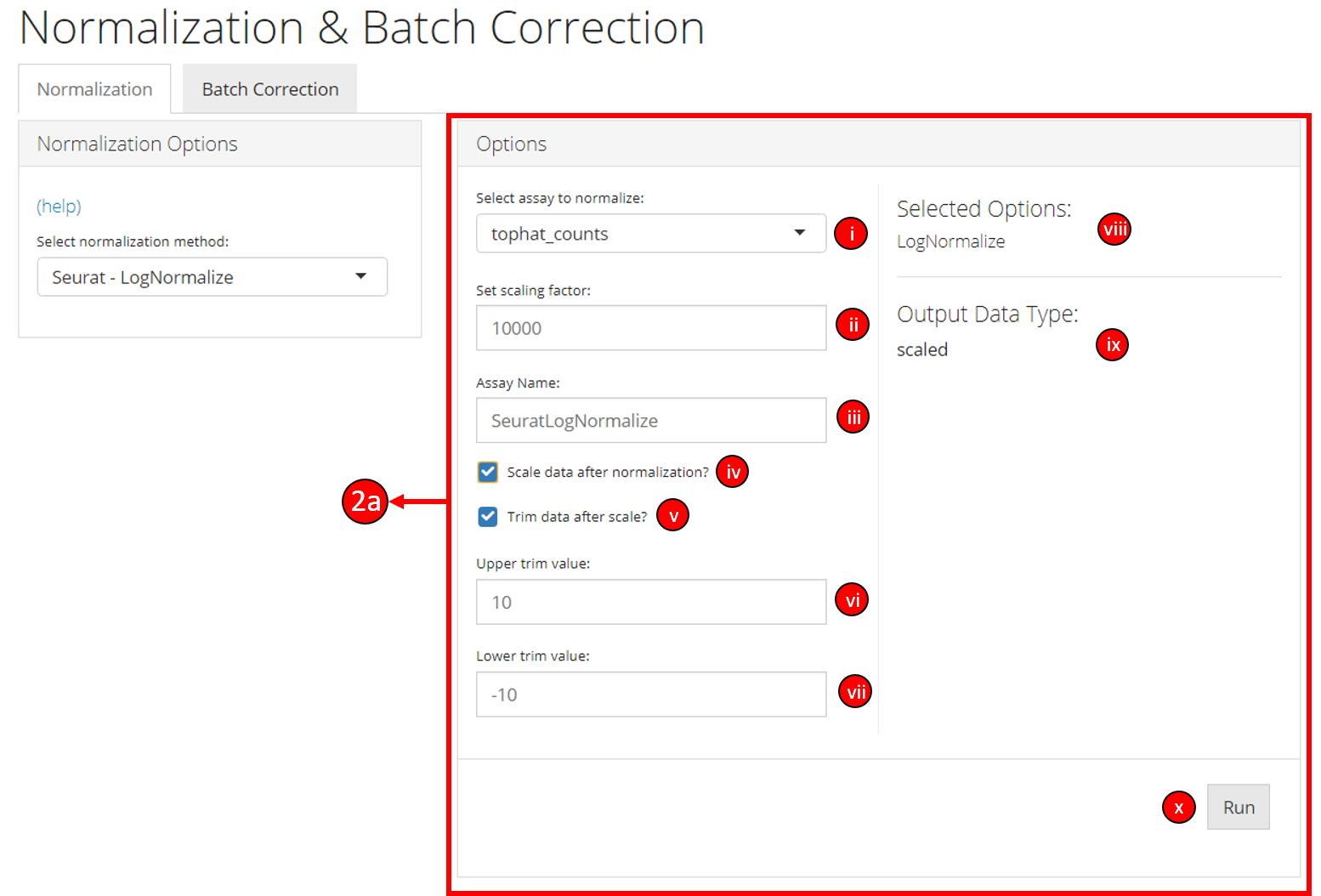
2a. Options when a normalization method from a
particular package is selected:
i. Select the assay from the data
to normalize.
ii. Set scaling factor for this particular method.
These options may vary between different normalization methods. See
method specific documentation for more information.
iii. Specify
the name of the output data assay.
iv. Select if data should be
scaled after running the normalization method.
v. Select if data
should be trimmed between two values running the normalization method.
vi & vii. If trim data option is selected, specify the upper
and lower trim values. The output data is then trimmed between these two
values (inclusive).
viii: The user-interface displays the selected
options or methods based on the user selected normalization method.
ix. The output data type based on the selected user options is
displayed.
x. Click the button to run the normalization process.
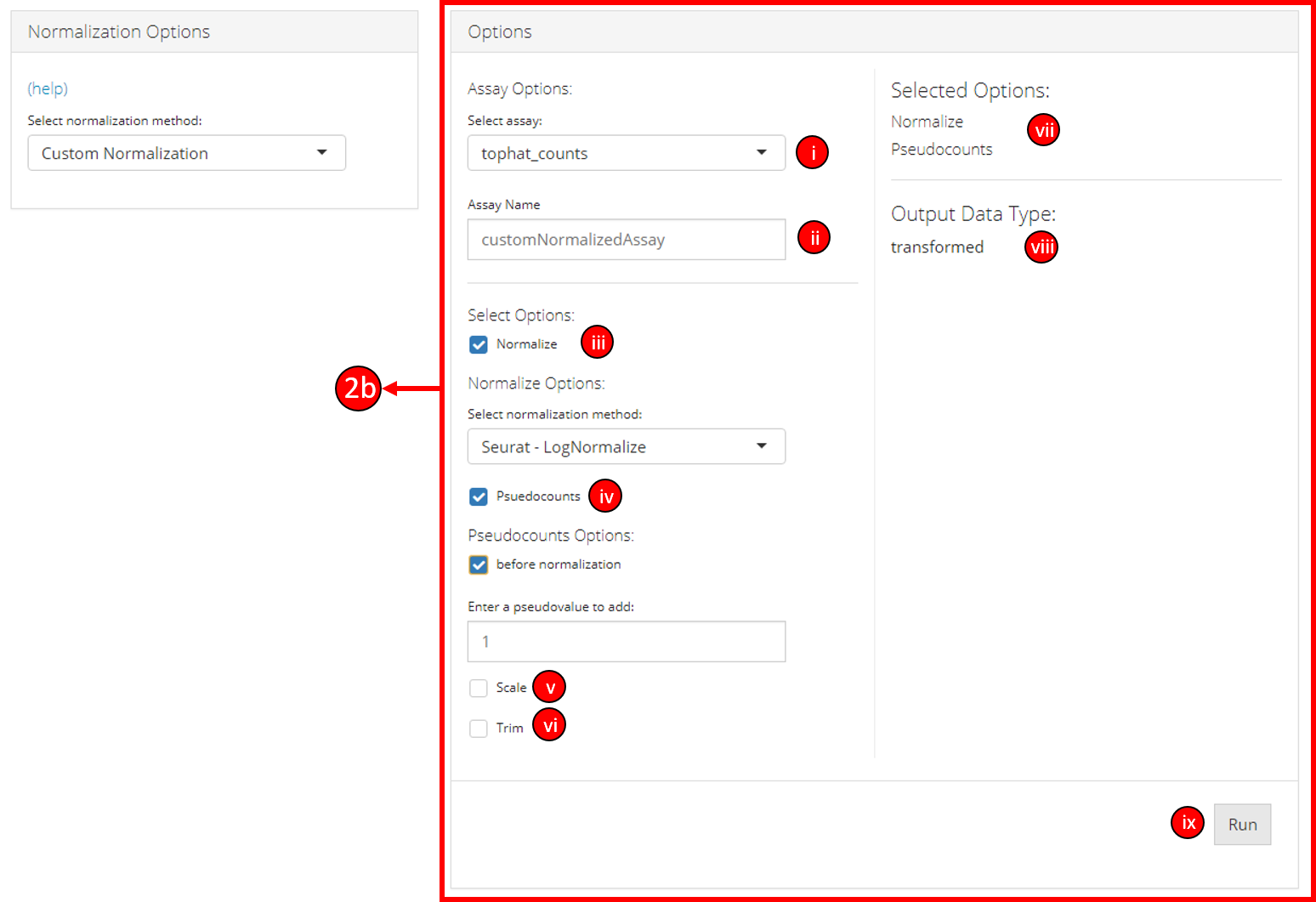
2b. Options when a custom normalization is selected:
i. Select the input data assay to use as an input with selected
normalization/transformation options.
ii. Specify the name of the
put data assay.
iii. Select if data should be normalized (multiple
normalization methods will display once this option is selected).
iv. Select if a pseudo value should be added to the data.
v. Select
if the data should be scaled.
vi. Select if the data should be
trimmed between two values.
vii. The user-interface displays the
selected options or methods based on the user selected normalization
method.
viii. The output data type based on the selected user
options is displayed.
ix. Click the button to run the normalization
process.
The singleCellTK allows the users to run all
normalization and transformation methods on the input data by using a
single runNormalization function. The
runNormalization function takes in input a
SingleCellExperiment object and a series of parameters that
define the normalization/transformation options to run on the specified
assay. The output of this function is a
SingleCellExperiment object which now contains the
normalized/transformed assay.
To use the function, input a SingleCellExperiment object
that contains the data assay and specify the required parameters:
sce <- runNormalization(inSCE = sce, normalizationMethod = "RC", useAssay = "counts", outAssayName = "RCLogScaledCounts", scale = TRUE, transformation = "log2", pseudocountsBeforeTransform = 1, trim = c(10, -10))Example
# Load singleCellTK & pbmc3k example data
library(singleCellTK)
sce <- importExampleData(dataset = "pbmc3k")
# Perform normalization
sce <- runNormalization(inSCE = sce, normalizationMethod = "RC", useAssay = "counts", outAssayName = "RCLogScaledCounts", scale = TRUE, transformation = "log2", pseudocountsBeforeTransform = 1, trim = c(10, -10))Parameters
The runNormalization function specifies the following
parameters:
| Parameter | Description |
|---|---|
| inSCE | Input SingleCellExperiment object. |
| useAssay | Specify the input assay to use for
normalization/transformation. |
| outAssayName | A character value indicating the name of the new output
assay. |
| normalizationMethod | Specify a normalization method from "LogNormalize",
"CLR", "RC", "SCTransform",
"logNormCounts" or "CPM". If no method is
specified, normalization will not be performed. |
| scale | Logical value indicating if Z.Score scaling should be performed or not. |
| seuratScaleFactor | Specify the scaleFactor parameter if any of the
seurat normalization method is selected. |
| transformation | Specify if a transformation should be applied to the input
assay (if normalization is selected, this transformation is
applied after normalization). Available transformation options include
"log2", "log1p", "sqrt". |
| pseudocountsBeforeNorm | A numeric value to add to the input assay
before performing normalization. |
| pseudocountsBeforeTransform | A numeric value to add to the input assay
before performing a transformation. |
| trim | A numeric(2) vector that specifies the upper and the
lower trim values between (exclusive) which the input assay
should be trimmed. |
| verbose | A logical value indicating if informative/progress
messages should be displayed on the console. |
Individual Functions
While the runNormalization wrapper function can be used
to perform all available normalization and transformation options,
separate functions are also available for all of the included
normalization methods. The following functions can be used for specific
normalization methods:
LogNormalize, CLR or RC from
Seurat [[1]][2] package:
sce <- runSeuratNormalizeData(inSCE = sce, normalizationMethod = "CLR", useAssay = "counts", normAssayName = "CLRCounts", scaleFactor = 10000, verbose = TRUE)The parameters to the above function include: inSCE: an input SingleCellExperiment object
normalizationMethod: one of the options from
“LogNormalize”, “CLR” or “RC” useAssay: name of the
assay to use for normalization normAssayName: name of
the output normalized assay scaleFactor: a numeric
value that represents the scaling factor verbose: a
logical value indicating if progress should be printed
SCTransform from Seurat [[1]][2][6] package:
sce <- runSeuratSCTransform(inSCE = sce, useAssay = "counts", normAssayName = "SCTCounts", verbose = TRUE)The parameters to the above function include: inSCE: an input SingleCellExperiment object
useAssay: name of the assay to use for normalization
normAssayName: name of the output normalized assay
verbose: a logical value indicating if progress should
be printed
logNormCounts from Scater [5] package:
sce <- scaterlogNormCounts(inSCE = sce, useAssay = "counts", assayName = "logcounts")The parameters to the above function include: inSCE: an input SingleCellExperiment object
useAssay: name of the assay to use for normalization
assayName: name of the output normalized assay
CPM from Scater [5] package:
sce <- scaterCPM(inSCE = sce, useAssay = "counts", assayName = "countsCPM")The parameters to the above function include: inSCE: an input SingleCellExperiment object
useAssay: name of the assay to use for normalization
assayName: name of the output normalized assay
Compute Z-Score:
assay(sce, "countsZScore") <- computeZScore(counts = assay(sce, "counts"))The parameters to the above function include: counts: input matrix to scale with Z-Score
Matrix trimming:
assay(sce, "countsTrimmed") <- trimCounts(counts = assay(sce, "counts"), trimValue = c(10, -10))The parameters to the above function include: counts: an input SingleCellExperiment object
trimValue: a numeric(2) vector with the
first value indicating the upper trim value and the second value
indicating the lower trim value