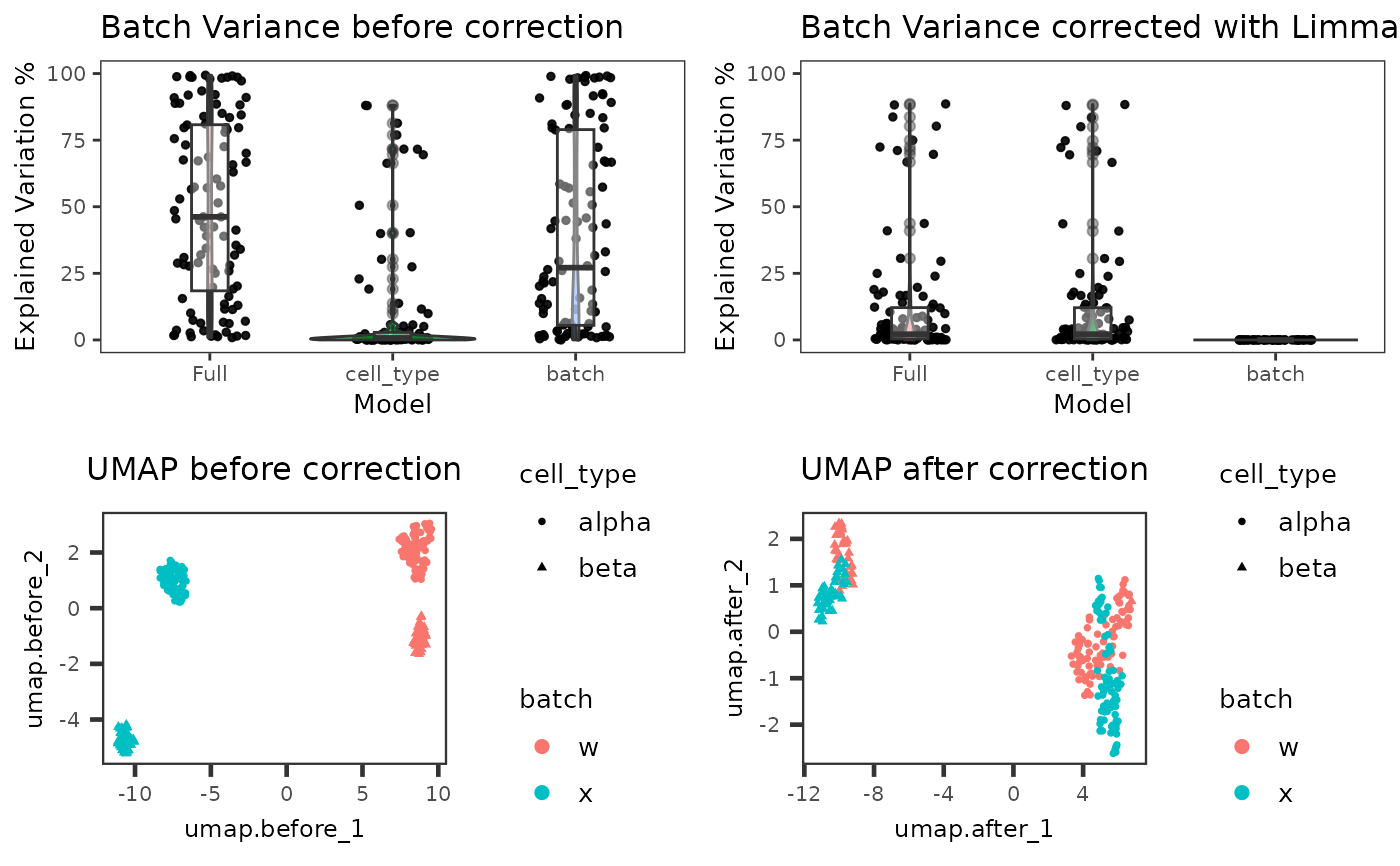
Plot comparison of batch corrected result against original assay
Source:R/plotBatchVariance.R
plotBatchCorrCompare.RdPlot comparison of batch corrected result against original assay
plotBatchCorrCompare(
inSCE,
corrMat,
batch = NULL,
condition = NULL,
origAssay = NULL,
origLogged = NULL,
method = NULL,
matType = NULL
)Arguments
- inSCE
SingleCellExperiment inherited object.
- corrMat
A single character indicating the name of the corrected matrix.
- batch
A single character. The name of batch annotation column in
colData(inSCE).- condition
A single character. The name of an additional covariate annotation column in
colData(inSCE).- origAssay
A single character indicating what the original assay used for batch correction is.
- origLogged
Logical scalar indicating whether
origAssayis log-normalized.- method
A single character indicating the name of the batch correction method. Only used for the titles of plots.
- matType
A single character indicating the type of the batch correction result matrix, choose from
"assay","altExp","reducedDim".
Value
An object of class "gtable", combining four ggplots.
Details
Four plots will be combined. Two of them are violin/box-plots for percent variance explained by the batch variation, and optionally the covariate, for original and corrected. The other two are UMAPs of the original assay and the correction result matrix. If SCTK batch correction methods are performed in advance, this function will automatically detect necessary input. Otherwise, users can also customize the input. Future improvement might include solution to reduce redundant UMAP calculation.
Examples
data("sceBatches")
logcounts(sceBatches) <- log1p(counts(sceBatches))
sceBatches <- runLimmaBC(sceBatches)
plotBatchCorrCompare(sceBatches, "LIMMA", condition = "cell_type")
#> Sat Mar 18 10:28:21 2023 ... Computing Scater UMAP for sample '1'.
#> Sat Mar 18 10:28:24 2023 ... Computing Scater UMAP for sample '1'.
#> Warning: Removed 47 rows containing missing values (`geom_point()`).