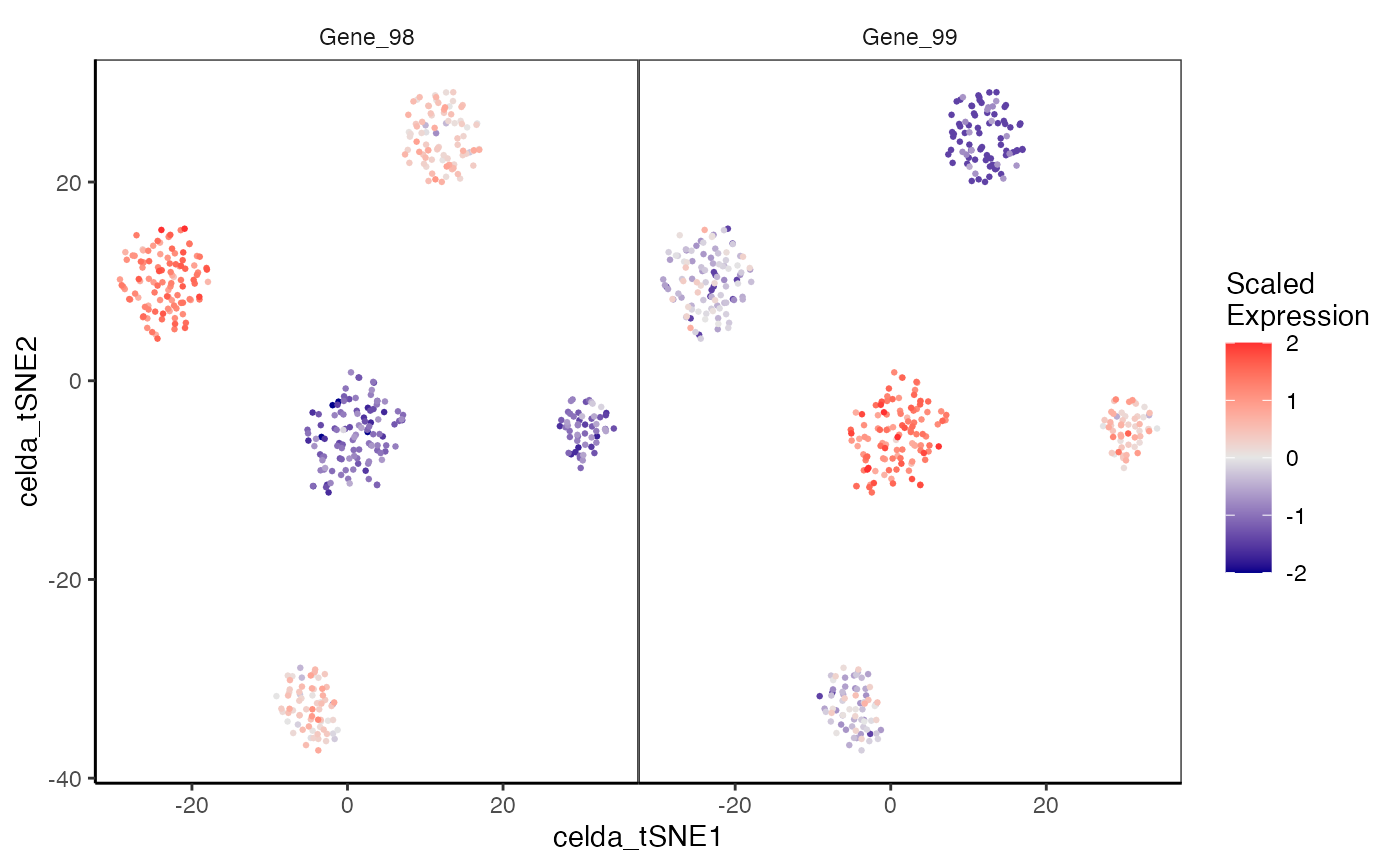
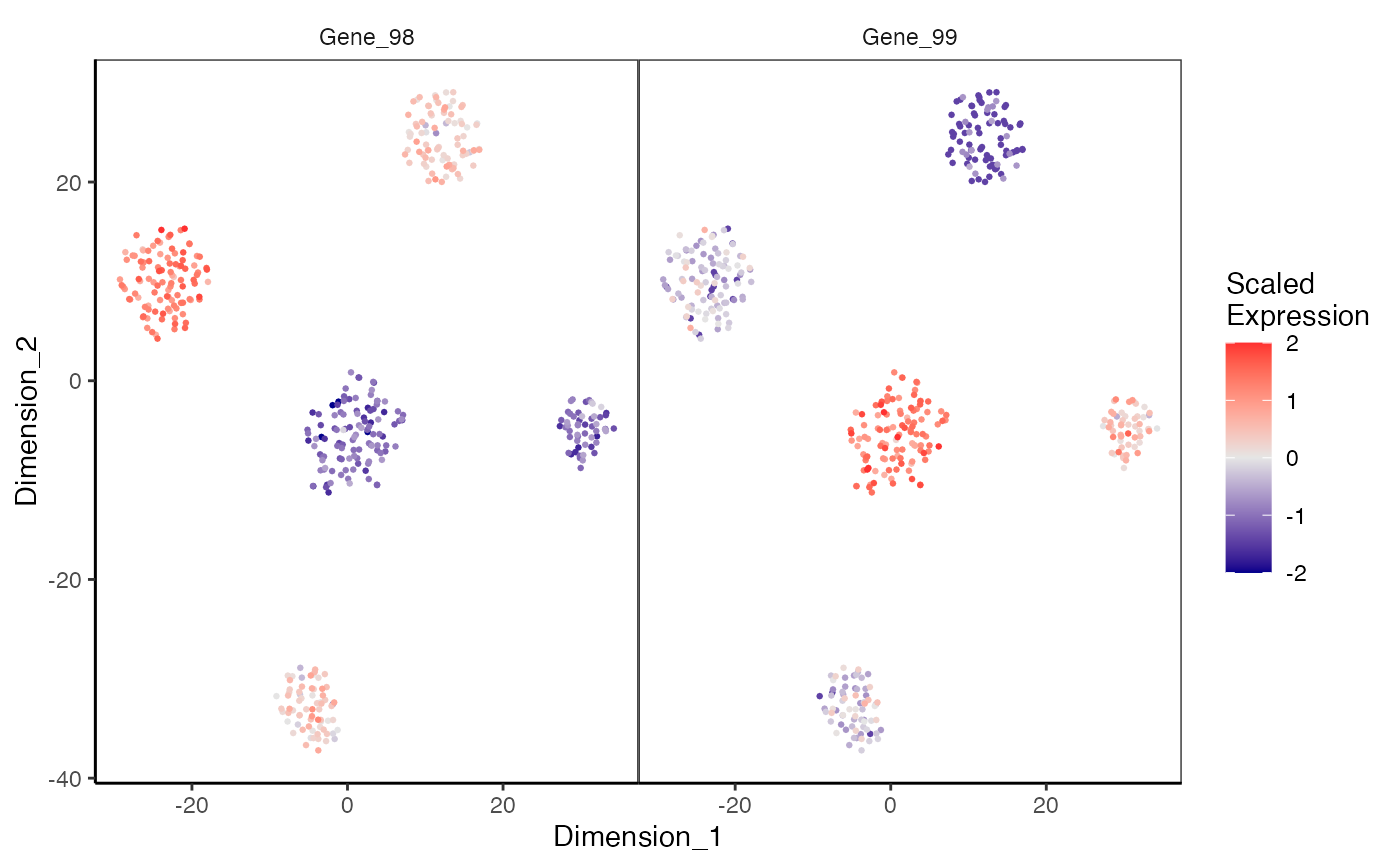
Plotting feature expression on a dimension reduction plot
Source:R/plot_dr.R
plotDimReduceFeature.RdCreate a scatterplot for each row of a normalized gene expression matrix where x and y axis are from a data dimension reduction tool. The cells are colored by expression of the specified feature.
plotDimReduceFeature( x, features, reducedDimName = NULL, displayName = NULL, dim1 = NULL, dim2 = NULL, headers = NULL, useAssay = "counts", altExpName = "featureSubset", normalize = FALSE, zscore = TRUE, exactMatch = TRUE, trim = c(-2, 2), limits = c(-2, 2), size = 0.5, xlab = NULL, ylab = NULL, colorLow = "blue4", colorMid = "grey90", colorHigh = "firebrick1", midpoint = 0, ncol = NULL, decreasing = FALSE ) # S4 method for SingleCellExperiment plotDimReduceFeature( x, features, reducedDimName = NULL, displayName = NULL, dim1 = 1, dim2 = 2, headers = NULL, useAssay = "counts", altExpName = "featureSubset", normalize = FALSE, zscore = TRUE, exactMatch = TRUE, trim = c(-2, 2), limits = c(-2, 2), size = 0.5, xlab = NULL, ylab = NULL, colorLow = "blue4", colorMid = "grey90", colorHigh = "firebrick1", midpoint = 0, ncol = NULL, decreasing = FALSE ) # S4 method for ANY plotDimReduceFeature( x, features, dim1, dim2, headers = NULL, normalize = FALSE, zscore = TRUE, exactMatch = TRUE, trim = c(-2, 2), limits = c(-2, 2), size = 0.5, xlab = "Dimension_1", ylab = "Dimension_2", colorLow = "blue4", colorMid = "grey90", colorHigh = "firebrick1", midpoint = 0, ncol = NULL, decreasing = FALSE )
Arguments
| x | Numeric matrix or a SingleCellExperiment object
with the matrix located in the assay slot under |
|---|---|
| features | Character vector. Features in the rownames of counts to plot. |
| reducedDimName | The name of the dimension reduction slot in
|
| displayName | Character. The column name of
|
| dim1 | Integer or numeric vector. If |
| dim2 | Integer or numeric vector. If |
| headers | Character vector. If |
| useAssay | A string specifying which assay
slot to use if |
| altExpName | The name for the altExp slot to use. Default "featureSubset". |
| normalize | Logical. Whether to normalize the columns of `counts`.
Default |
| zscore | Logical. Whether to scale each feature to have a mean 0
and standard deviation of 1. Default |
| exactMatch | Logical. Whether an exact match or a partial match using
|
| trim | Numeric vector. Vector of length two that specifies the lower
and upper bounds for the data. This threshold is applied after row scaling.
Set to NULL to disable. Default |
| limits | Passed to scale_colour_gradient2. The range of color scale. |
| size | Numeric. Sets size of point on plot. Default 1. |
| xlab | Character vector. Label for the x-axis. If |
| ylab | Character vector. Label for the y-axis. If |
| colorLow | Character. A color available from `colors()`. The color will be used to signify the lowest values on the scale. |
| colorMid | Character. A color available from `colors()`. The color will be used to signify the midpoint on the scale. |
| colorHigh | Character. A color available from `colors()`. The color will be used to signify the highest values on the scale. |
| midpoint | Numeric. The value indicating the midpoint of the
diverging color scheme. If |
| ncol | Integer. Passed to facet_wrap. Specify the number of columns for facet wrap. |
| decreasing | logical. Specifies the order of plotting the points.
If |
Value
The plot as a ggplot object
Examples
data(sceCeldaCG) sce <- celdaTsne(sceCeldaCG) plotDimReduceFeature(x = sce, reducedDimName = "celda_tSNE", normalize = TRUE, features = c("Gene_98", "Gene_99"), exactMatch = TRUE)library(SingleCellExperiment) data(sceCeldaCG) sce <- celdaTsne(sceCeldaCG) plotDimReduceFeature(x = counts(sce), dim1 = reducedDim(altExp(sce), "celda_tSNE")[, 1], dim2 = reducedDim(altExp(sce), "celda_tSNE")[, 2], normalize = TRUE, features = c("Gene_98", "Gene_99"), exactMatch = TRUE)